
Whole-Genome Comparative Phylogeography
Phylogeography is the study of population genetic variation in a geographic context. Sequence data contains a record of the history of a species and we can use statistical tools to make inference about this history. Comparative phylogeography extends this idea to investigate whether multiple co-distributed species have undergone similar population dynamics (e.g. bottenecks, expansions, populations splits) given the fact that they were historically subject to the same environmental conditions.
Recently, collaborators and I have developed a comparative phylogeographic method for whole-genome data. We call this method Phylogeograpic Temporal Analysis (PTA) A couple figures to demonstrate the core concepts:
Do shared demographic histories create similar patterns in the genetic data?
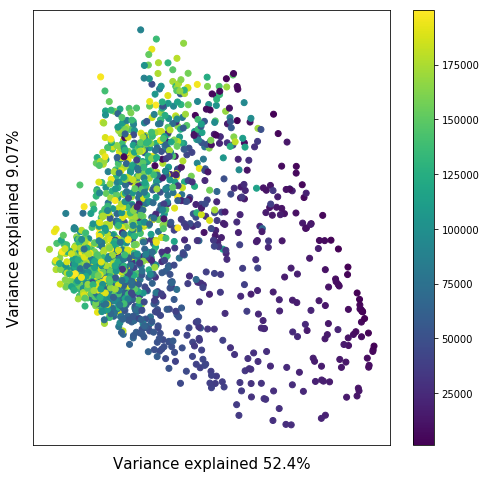
This figure depicts a principal component dimension-reduction of 2000 independent mSFS (multi-site frequency spectra) generated by simulating 10 co-expanding populations with identical co-expansion magnitudes, while only allowing the time of co-expansion to vary. Each point is a simulation, and the color of the point indicates the time of co-expansion. The figure indicates that there are observable differences between the mSFS generated by assemblages co-expanding at different times.
Can we use machine learning to estimate model parameters relevant to shared demographic histories?
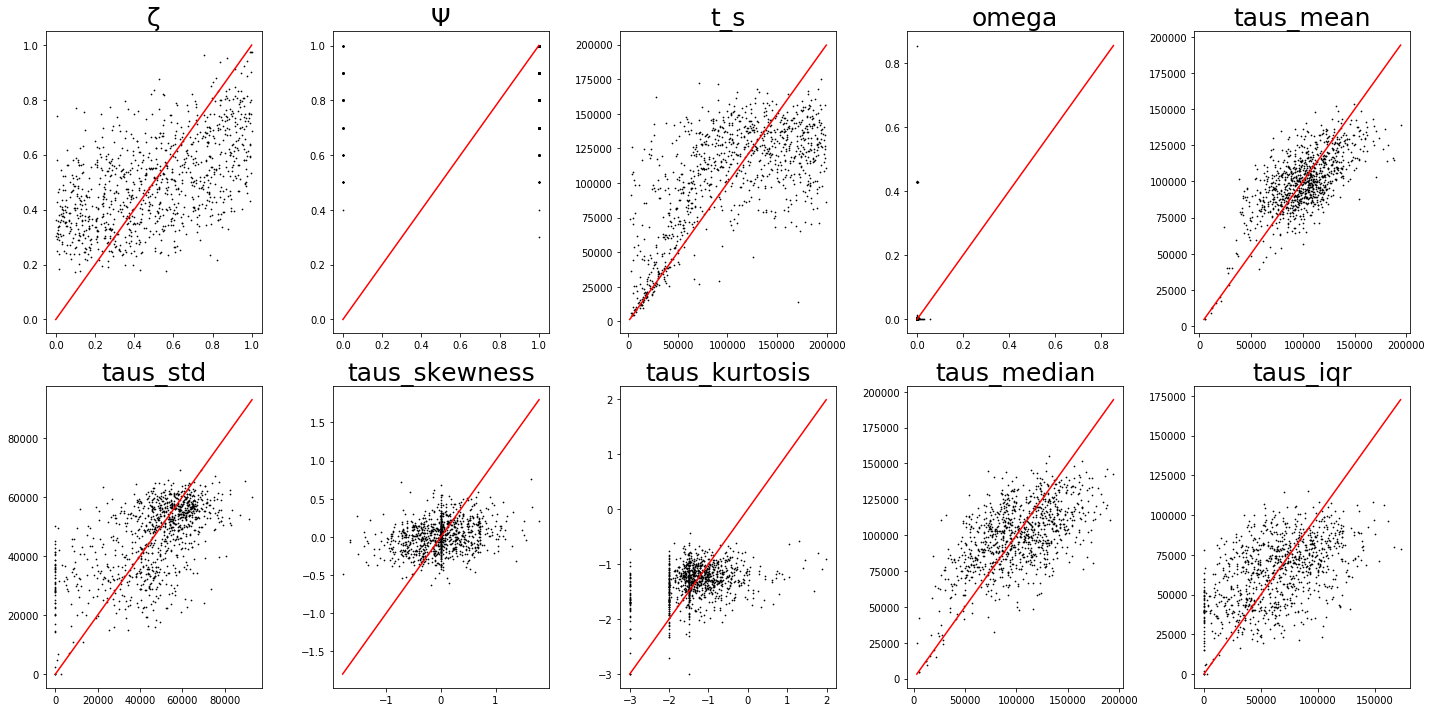
Here we see the results of a typical ML regression cross-validation procedure. Simulations are used to train the ML regressor and then held-out datasets (generated with known parameter values) are presented to the ML model with the goal of testing accuracy of prediction of the known parameter values. Hi-fidelity ML inference should produce accurate predictions, and the cloud of points should fall on the identity line.
Again, this is early development stage, so even modest correlations between true and predicted values indicate the potential of the ML framework. Given further testing and development cross-validation results will only continue to improve.
The future of comparative phylogeography
Comparative phylogeographic methods were first developed for single locus datasets, typically several hundred basepairs of a single mitochondrial marker. Given that single locus data can give a biased picture of population history, methods have advanced through time to support multi-locus data, and even SNP data representing thousands of independent loci across the genome, typical of RADSeq experiments.
PTA is the only comparative phylogeographic method that can make use of the information in whole-genome sequences, while fully accounting for linkage. We also designed it to have a simple, modern interface, a massively parallel backend, and an automated machine learning inference procedure. PTA is the most advanced comparative phylogeographic method developed to date, and we’re just getting started!